Where Do the Instructions for Building a Protein Come From Originally?
In the preceding section we have seen that the final product of some genes is an RNA molecule itself, such as those present in the snRNPs and in ribosomes. However, most genes in a cell produce mRNA molecules that serve as intermediaries on the pathway to proteins. In this section we examine how the cell converts the information carried in an mRNA molecule into a protein molecule. This feat of translation first attracted the attention of biologists in the late 1950s, when it was posed as the "coding problem": how is the information in a linear sequence of nucleotides in RNA translated into the linear sequence of a chemically quite different set of subunits—the amino acids in proteins? This fascinating question stimulated great excitement among scientists at the time. Here was a cryptogram set up by nature that, after more than 3 billion years of evolution, could finally be solved by one of the products of evolution—human beings. And indeed, not only has the code been cracked step by step, but in the year 2000 the elaborate machinery by which cells read this code—the ribosome—was finally revealed in atomic detail.
An mRNA Sequence Is Decoded in Sets of Three Nucleotides
Once an mRNA has been produced, by transcription and processing the information present in its nucleotide sequence is used to synthesize a protein. Transcription is simple to understand as a means of information transfer: since DNA and RNA are chemically and structurally similar, the DNA can act as a direct template for the synthesis of RNA by complementary base-pairing. As the term transcription signifies, it is as if a message written out by hand is being converted, say, into a typewritten text. The language itself and the form of the message do not change, and the symbols used are closely related.
In contrast, the conversion of the information in RNA into protein represents a translation of the information into another language that uses quite different symbols. Moreover, since there are only four different nucleotides in mRNA and twenty different types of amino acids in a protein, this translation cannot be accounted for by a direct one-to-one correspondence between a nucleotide in RNA and an amino acid in protein. The nucleotide sequence of a gene, through the medium of mRNA, is translated into the amino acid sequence of a protein by rules that are known as the genetic code. This code was deciphered in the early 1960s.
The sequence of nucleotides in the mRNA molecule is read consecutively in groups of three. RNA is a linear polymer of four different nucleotides, so there are 4 × 4 × 4 = 64 possible combinations of three nucleotides: the triplets AAA, AUA, AUG, and so on. However, only 20 different amino acids are commonly found in proteins. Either some nucleotide triplets are never used, or the code is redundant and some amino acids are specified by more than one triplet. The second possibility is, in fact, the correct one, as shown by the completely deciphered genetic code in Figure 6-50. Each group of three consecutive nucleotides in RNA is called a codon, and each codon specifies either one amino acid or a stop to the translation process.

Figure 6-50
The genetic code. The standard one-letter abbreviation for each amino acid is presented below its three-letter abbreviation (see Panel 3-1, pp. 132–133, for the full name of each amino acid and its structure). By convention, codons are always (more...)
This genetic code is used universally in all present-day organisms. Although a few slight differences in the code have been found, these are chiefly in the DNA of mitochondria. Mitochondria have their own transcription and protein synthesis systems that operate quite independently from those of the rest of the cell, and it is understandable that their small genomes have been able to accommodate minor changes to the code (discussed in Chapter 14).
In principle, an RNA sequence can be translated in any one of three different reading frames, depending on where the decoding process begins (Figure 6-51). However, only one of the three possible reading frames in an mRNA encodes the required protein. We see later how a special punctuation signal at the beginning of each RNA message sets the correct reading frame at the start of protein synthesis.

Figure 6-51
The three possible reading frames in protein synthesis. In the process of translating a nucleotide sequence (blue) into an amino acid sequence (green), the sequence of nucleotides in an mRNA molecule is read from the 5′ to the 3′ end in (more...)
tRNA Molecules Match Amino Acids to Codons in mRNA
The codons in an mRNA molecule do not directly recognize the amino acids they specify: the group of three nucleotides does not, for example, bind directly to the amino acid. Rather, the translation of mRNA into protein depends on adaptor molecules that can recognize and bind both to the codon and, at another site on their surface, to the amino acid. These adaptors consist of a set of small RNA molecules known as transfer RNAs (tRNAs), each about 80 nucleotides in length.
We saw earlier in this chapter that RNA molecules can fold up into precisely defined three-dimensional structures, and the tRNA molecules provide a striking example. Four short segments of the folded tRNA are double-helical, producing a molecule that looks like a cloverleaf when drawn schematically (Figure 6-52A). For example, a 5′-GCUC-3′ sequence in one part of a polynucleotide chain can form a relatively strong association with a 5′-GAGC-3′ sequence in another region of the same molecule. The cloverleaf undergoes further folding to form a compact L-shaped structure that is held together by additional hydrogen bonds between different regions of the molecule (Figure 6-52B,C).
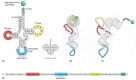
Figure 6-52
A tRNA molecule. In this series of diagrams, the same tRNA molecule—in this case a tRNA specific for the amino acid phenylalanine (Phe)—is depicted in various ways. (A) The cloverleaf structure, a convention used to show the complementary (more...)
Two regions of unpaired nucleotides situated at either end of the L-shaped molecule are crucial to the function of tRNA in protein synthesis. One of these regions forms the anticodon, a set of three consecutive nucleotides that pairs with the complementary codon in an mRNA molecule. The other is a short single-stranded region at the 3′ end of the molecule; this is the site where the amino acid that matches the codon is attached to the tRNA.
We have seen in the previous section that the genetic code is redundant; that is, several different codons can specify a single amino acid (see Figure 6-50). This redundancy implies either that there is more than one tRNA for many of the amino acids or that some tRNA molecules can base-pair with more than one codon. In fact, both situations occur. Some amino acids have more than one tRNA and some tRNAs are constructed so that they require accurate base-pairing only at the first two positions of the codon and can tolerate a mismatch (or wobble) at the third position (Figure 6-53). This wobble base-pairing explains why so many of the alternative codons for an amino acid differ only in their third nucleotide (see Figure 6-50). In bacteria, wobble base-pairings make it possible to fit the 20 amino acids to their 61 codons with as few as 31 kinds of tRNA molecules. The exact number of different kinds of tRNAs, however, differs from one species to the next. For example, humans have 497 tRNA genes but, among them, only 48 different anticodons are represented.

Figure 6-53
Wobble base-pairing between codons and anticodons. If the nucleotide listed in the first column is present at the third, or wobble, position of the codon, it can base-pair with any of the nucleotides listed in the second column. Thus, for example, when (more...)
tRNAs Are Covalently Modified Before They Exit from the Nucleus
We have seen that most eucaryotic RNAs are covalently altered before they are allowed to exit from the nucleus, and tRNAs are no exception. Eucaryotic tRNAs are synthesized by RNA polymerase III. Both bacterial and eucaryotic tRNAs are typically synthesized as larger precursor tRNAs, and these are then trimmed to produce the mature tRNA. In addition, some tRNA precursors (from both bacteria and eucaryotes) contain introns that must be spliced out. This splicing reaction is chemically distinct from that of pre-mRNA splicing; rather than generating a lariat intermediate, tRNA splicing occurs through a cut-and-paste mechanism that is catalyzed by proteins (Figure 6-54). Trimming and splicing both require the precursor tRNA to be correctly folded in its cloverleaf configuration. Because misfolded tRNA precursors will not be processed properly, the trimming and splicing reactions are thought to act as quality-control steps in the generation of tRNAs.

Figure 6-54
Structure of a tRNA-splicing endonuclease docked to a precursor tRNA. The endonuclease (a four-subunit enzyme) removes the tRNA intron (blue). A second enzyme, a multifunctional tRNA ligase (not shown), then joins the two tRNA halves together. (Courtesy (more...)
All tRNAs are also subject to a variety of chemical modifications—nearly one in 10 nucleotides in each mature tRNA molecule is an altered version of a standard G, U, C, or A ribonucleotide. Over 50 different types of tRNA modifications are known; a few are shown in Figure 6-55. Some of the modified nucleotides—most notably inosine, produced by the deamination of guanosine—affect the conformation and base-pairing of the anticodon and thereby facilitate the recognition of the appropriate mRNA codon by the tRNA molecule (see Figure 6-53). Others affect the accuracy with which the tRNA is attached to the correct amino acid.
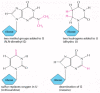
Figure 6-55
A few of the unusual nucleotides found in tRNA molecules. These nucleotides are produced by covalent modification of a normal nucleotide after it has been incorporated into an RNA chain. In most tRNA molecules about 10% of the nucleotides are modified (more...)
Specific Enzymes Couple Each Amino Acid to Its Appropriate tRNA Molecule
We have seen that, to read the genetic code in DNA, cells make a series of different tRNAs. We now consider how each tRNA molecule becomes linked to the one amino acid in 20 that is its appropriate partner. Recognition and attachment of the correct amino acid depends on enzymes called aminoacyl-tRNA synthetases, which covalently couple each amino acid to its appropriate set of tRNA molecules (Figures 6-56 and 6-57). For most cells there is a different synthetase enzyme for each amino acid (that is, 20 synthetases in all); one attaches glycine to all tRNAs that recognize codons for glycine, another attaches alanine to all tRNAs that recognize codons for alanine, and so on. Many bacteria, however, have fewer than 20 synthetases, and the same synthetase enzyme is responsible for coupling more than one amino acid to the appropriate tRNAs. In these cases, a single synthetase places the identical amino acid on two different types of tRNAs, only one of which has an anticodon that matches the amino acid. A second enzyme then chemically modifies each "incorrectly" attached amino acid so that it now corresponds to the anticodon displayed by its covalently linked tRNA.
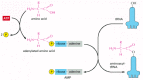
Figure 6-56
Amino acid activation. The two-step process in which an amino acid (with its side chain denoted by R) is activated for protein synthesis by an aminoacyl-tRNA synthetase enzyme is shown. As indicated, the energy of ATP hydrolysis is used to attach each (more...)
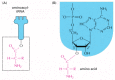
Figure 6-57
The structure of the aminoacyl-tRNA linkage. The carboxyl end of the amino acid forms an ester bond to ribose. Because the hydrolysis of this ester bond is associated with a large favorable change in free energy, an amino acid held in this way is said (more...)
The synthetase-catalyzed reaction that attaches the amino acid to the 3′ end of the tRNA is one of many cellular reactions coupled to the energy-releasing hydrolysis of ATP (see pp. 83–84), and it produces a high-energy bond between the tRNA and the amino acid. The energy of this bond is used at a later stage in protein synthesis to link the amino acid covalently to the growing polypeptide chain.
Although the tRNA molecules serve as the final adaptors in converting nucleotide sequences into amino acid sequences, the aminoacyl-tRNA synthetase enzymes are adaptors of equal importance in the decoding process (Figure 6-58). This was established by an ingenious experiment in which an amino acid (cysteine) was chemically converted into a different amino acid (alanine) after it already had been attached to its specific tRNA. When such "hybrid" aminoacyl-tRNA molecules were used for protein synthesis in a cell-free system, the wrong amino acid was inserted at every point in the protein chain where that tRNA was used. Although cells have several quality control mechanisms to avoid this type of mishap, the experiment clearly establishes that the genetic code is translated by two sets of adaptors that act sequentially. Each matches one molecular surface to another with great specificity, and it is their combined action that associates each sequence of three nucleotides in the mRNA mole-cule—that is, each codon—with its particular amino acid.

Figure 6-58
The genetic code is translated by means of two adaptors that act one after another. The first adaptor is the aminoacyl-tRNA synthetase, which couples a particular amino acid to its corresponding tRNA; the second adaptor is the tRNA molecule itself, whose (more...)
Editing by RNA Synthetases Ensures Accuracy
Several mechanisms working together ensure that the tRNA synthetase links the correct amino acid to each tRNA. The synthetase must first select the correct amino acid, and most do so by a two-step mechanism. First, the correct amino acid has the highest affinity for the active-site pocket of its synthetase and is therefore favored over the other 19. In particular, amino acids larger than the correct one are effectively excluded from the active site. However, accurate discrimination between two similar amino acids, such as isoleucine and valine (which differ by only a methyl group), is very difficult to achieve by a one-step recognition mechanism. A second discrimination step occurs after the amino acid has been covalently linked to AMP (see Figure 6-56). When tRNA binds the synthetase, it forces the amino acid into a second pocket in the synthetase, the precise dimensions of which exclude the correct amino acid but allow access by closely related amino acids. Once an amino acid enters this editing pocket, it is hydrolyzed from the AMP (or from the tRNA itself if the aminoacyl-tRNA bond has already formed) and released from the enzyme. This hydrolytic editing, which is analogous to the editing by DNA polymerases (Figure 6-59), raises the overall accuracy of tRNA charging to approximately one mistake in 40,000 couplings.
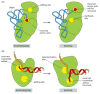
Figure 6-59
Hydrolytic editing. (A) tRNA synthetases remove their own coupling errors through hydrolytic editing of incorrectly attached amino acids. As described in the text, the correct amino acid is rejected by the editing site. (B) The error-correction process (more...)
The tRNA synthetase must also recognize the correct set of tRNAs, and extensive structural and chemical complementarity between the synthetase and the tRNA allows various features of the tRNA to be sensed (Figure 6-60). Most tRNA synthetases directly recognize the matching tRNA anticodon; these synthetases contain three adjacent nucleotide-binding pockets, each of which is complementary in shape and charge to the nucleotide in the anticodon. For other synthetases it is the nucleotide sequence of the acceptor stem that is the key recognition determinant. In most cases, however, nucleotides at several positions on the tRNA are "read" by the synthetase.

Figure 6-60
The recognition of a tRNA molecule by its aminoacyl-tRNA synthetase. For this tRNA (tRNAGln), specific nucleotides in both the anticodon (bottom) and the amino acid-accepting arm allow the correct tRNA to be recognized by the synthetase enzyme (blue). (more...)
Amino Acids Are Added to the C-terminal End of a Growing Polypeptide Chain
Having seen that amino acids are first coupled to tRNA molecules, we now turn to the mechanism by which they are joined together to form proteins. The fundamental reaction of protein synthesis is the formation of a peptide bond between the carboxyl group at the end of a growing polypeptide chain and a free amino group on an incoming amino acid. Consequently, a protein is synthesized stepwise from its N-terminal end to its C-terminal end. Throughout the entire process the growing carboxyl end of the polypeptide chain remains activated by its covalent attachment to a tRNA molecule (a peptidyl-tRNA molecule). This high-energy covalent linkage is disrupted during each addition but is immediately replaced by the identical linkage on the most recently added amino acid (Figure 6-61). In this way, each amino acid added carries with it the activation energy for the addition of the next amino acid rather than the energy for its own addition—an example of the "head growth" type of polymerization described in Figure 2-68.

Figure 6-61
The incorporation of an amino acid into a protein. A polypeptide chain grows by the stepwise addition of amino acids to its C-terminal end. The formation of each peptide bond is energetically favorable because the growing C-terminus has been activated (more...)
The RNA Message Is Decoded on Ribosomes
As we have seen, the synthesis of proteins is guided by information carried by mRNA molecules. To maintain the correct reading frame and to ensure accuracy (about 1 mistake every 10,000 amino acids), protein synthesis is performed in the ribosome, a complex catalytic machine made from more than 50 different proteins (the ribosomal proteins) and several RNA molecules, the ribosomal RNAs (rRNAs). A typical eucaryotic cell contains millions of ribosomes in its cytoplasm (Figure 6-62). As we have seen, eucaryotic ribosomal subunits are assembled at the nucleolus, by the association of newly transcribed and modified rRNAs with ribosomal proteins, which have been transported into the nucleus after their synthesis in the cytoplasm. The two ribosomal subunits are then exported to the cytoplasm, where they perform protein synthesis.
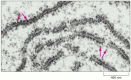
Figure 6-62
Ribosomes in the cytoplasm of a eucaryotic cell. This electron micrograph shows a thin section of a small region of cytoplasm. The ribosomes appear as black dots (red arrows). Some are free in the cytosol; others are attached to membranes of the endoplasmic (more...)
Eucaryotic and procaryotic ribosomes are very similar in design and function. Both are composed of one large and one small subunit that fit together to form a complete ribosome with a mass of several million daltons (Figure 6-63). The small subunit provides a framework on which the tRNAs can be accurately matched to the codons of the mRNA (see Figure 6-58), while the large subunit catalyzes the formation of the peptide bonds that link the amino acids together into a polypeptide chain (see Figure 6-61).
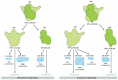
Figure 6-63
A comparison of the structures of procaryotic and eucaryotic ribosomes. Ribosomal components are commonly designated by their "S values," which refer to their rate of sedimentation in an ultracentrifuge. Despite the differences in the (more...)
When not actively synthesizing proteins, the two subunits of the ribosome are separate. They join together on an mRNA molecule, usually near its 5′ end, to initiate the synthesis of a protein. The mRNA is then pulled through the ribosome; as its codons encounter the ribosome's active site, the mRNA nucleotide sequence is translated into an amino acid sequence using the tRNAs as adaptors to add each amino acid in the correct sequence to the end of the growing polypeptide chain. When a stop codon is encountered, the ribosome releases the finished protein, its two subunits separate again. These subunits can then be used to start the synthesis of another protein on another mRNA molecule.
Ribosomes operate with remarkable efficiency: in one second, a single ribosome of a eucaryotic cell adds about 2 amino acids to a polypeptide chain; the ribosomes of bacterial cells operate even faster, at a rate of about 20 amino acids per second. How does the ribosome choreograph the many coordinated movements required for efficient translation? A ribosome contains four binding sites for RNA molecules: one is for the mRNA and three (called the A-site, the P-site, and the E-site) are for tRNAs (Figure 6-64). A tRNA molecule is held tightly at the A- and P-sites only if its anticodon forms base pairs with a complementary codon (allowing for wobble) on the mRNA molecule that is bound to the ribosome. The A- and P-sites are close enough together for their two tRNA molecules to be forced to form base pairs with adjacent codons on the mRNA molecule. This feature of the ribosome maintains the correct reading frame on the mRNA.

Figure 6-64
The RNA-binding sites in the ribosome. Each ribosome has three binding sites for tRNA: the A-, P-, and E-sites (short for aminoacyl-tRNA, peptidyl-tRNA, and exit, respectively) and one binding site for mRNA. (A) Structure of a bacterial ribosome with (more...)
Once protein synthesis has been initiated, each new amino acid is added to the elongating chain in a cycle of reactions containing three major steps. Our description of the chain elongation process begins at a point at which some amino acids have already been linked together and there is a tRNA molecule in the P-site on the ribosome, covalently joined to the end of the growing polypeptide (Figure 6-65). In step 1, a tRNA carrying the next amino acid in the chain binds to the ribosomal A-site by forming base pairs with the codon in mRNA positioned there, so that the P-site and the A-site contain adjacent bound tRNAs. In step 2, the carboxyl end of the polypeptide chain is released from the tRNA at the P-site (by breakage of the high-energy bond between the tRNA and its amino acid) and joined to the free amino group of the amino acid linked to the tRNA at the A-site, forming a new peptide bond. This central reaction of protein synthesis is catalyzed by a peptidyl transferase catalytic activity contained in the large ribosomal subunit. This reaction is accompanied by several conformational changes in the ribosome, which shift the two tRNAs into the E- and P-sites of the large subunit. In step 3, another series of conformational changes moves the mRNA exactly three nucleotides through the ribosome and resets the ribosome so it is ready to receive the next amino acyl tRNA. Step 1 is then repeated with a new incoming aminoacyl tRNA, and so on.
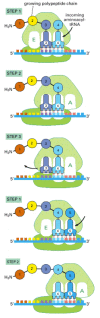
Figure 6-65
Translating an mRNA molecule. Each amino acid added to the growing end of a polypeptide chain is selected by complementary base-pairing between the anticodon on its attached tRNA molecule and the next codon on the mRNA chain. Because only one of the many (more...)
This three-step cycle is repeated each time an amino acid is added to the polypeptide chain, and the chain grows from its amino to its carboxyl end until a stop codon is encountered.
Elongation Factors Drive Translation Forward
The basic cycle of polypeptide elongation shown in outline in Figure 6-65 has an additional feature that makes translation especially efficient and accurate. Two elongation factors (EF-Tu and EF-G) enter and leave the ribosome during each cycle, each hydrolyzing GTP to GDP and undergoing conformational changes in the process. Under some conditions, ribosomes can be made to perform protein synthesis without the aid of the elongation factors and GTP hydrolysis, but this synthesis is very slow, inefficient, and inaccurate. The process is speeded up enormously by coupling conformational changes in the elongation factors to transitions between different conformational states of the ribosome. Although these conformational changes in the ribosome are not yet understood in detail, some may involve RNA rearrangements similar to those occurring in the RNAs of the spliceosome (see Figure 6-30). The cycles of elongation factor association, GTP hydrolysis, and dissociation ensures that the conformational changes occur in the "forward" direction and translation thereby proceeds efficiently (Figure 6-66).

Figure 6-66
Detailed view of the translation cycle. The outline of translation presented in Figure 6-65 has been supplemented with additional features, including the participation of elongation factors and a mechanism by which translational accuracy is improved. (more...)
In addition to helping move translation forward, EF-Tu is thought to increase the accuracy of translation by monitoring the initial interaction between a charged tRNA and a codon (see Figure 6-66). Charged tRNAs enter the ribosome bound to the GTP-form of EF-Tu. Although the bound elongation factor allows codon-anticodon pairing to occur, it prevents the amino acid from being incorporated into the growing polypeptide chain. The initial codon recognition, however, triggers the elongation factor to hydrolyze its bound GTP (to GDP and inorganic phosphate), whereupon the factor dissociates from the ribosome without its tRNA, allowing protein synthesis to proceed. The elongation factor introduces two short delays between codon-anticodon base pairing and polypeptide chain elongation; these delays selectively permit incorrectly bound tRNAs to exit from the ribosome before the irreversible step of chain elongation occurs. The first delay is the time required for GTP hydrolysis. The rate of GTP hydrolysis by EF-Tu is faster for a correct codon-anticodon pair than for an incorrect pair; hence an incorrectly bound tRNA molecule has a longer window of opportunity to dissociate from the ribosome. In other words, GTP hydrolysis selectively captures the correctly bound tRNAs. A second lag occurs between EF-Tu dissociation and the full accommodation of the tRNA in the A site of the ribosome. Although this lag is believed to be the same for correctly and incorrectly bound tRNAs, an incorrect tRNA molecule forms a smaller number of codon-anticodon hydrogen bonds than does a correctly matched pair and is therefore more likely to dissociate during this period. These two delays introduced by the elongation factor cause most incorrectly bound tRNA molecules (as well as a significant number of correctly bound molecules) to leave the ribosome without being used for protein synthesis, and this two-step mechanism is largely responsible for the 99.99% accuracy of the ribosome in translating proteins.
Recent discoveries indicate that EF-Tu may have an additional role in raising the overall accuracy of translation. Earlier in this chapter, we discussed the key role of aminoacyl synthetases in accurately matching amino acids to tRNAs. As the GTP-bound form of EF-Tu escorts aminoacyl-tRNAs to the ribosome (see Figure 6-66), it apparently double-checks for the proper correspondence between amino acid and tRNA and rejects those that are mismatched. Exactly how this is accomplished is not well-understood, but it may involve the overall binding energy between EF-Tu and the aminoacyl-tRNA. According to this idea, correct matches have a narrowly defined affinity for EF-Tu, and incorrect matches bind either too strongly or too weakly. EF-Tu thus appears to discriminate, albeit crudely, among many different amino acid-tRNA combinations, selectively allowing only the correct ones to enter the ribosome.
The Ribosome Is a Ribozyme
The ribosome is a very large and complex structure, composed of two-thirds RNA and one-third protein. The determination, in 2000, of the entire three-dimensional structure of its large and small subunits is a major triumph of modern structural biology. The structure strongly confirms the earlier evidence that rRNAs—and not proteins—are responsible for the ribosome's overall structure, its ability to position tRNAs on the mRNA, and its catalytic activity in forming covalent peptide bonds. Thus, for example, the ribosomal RNAs are folded into highly compact, precise three-dimensional structures that form the compact core of the ribosome and thereby determine its overall shape (Figure 6-67).

Figure 6-67
Structure of the rRNAs in the large subunit of a bacterial ribosome, as determined by x-ray crystallography. (A) Three-dimensional structures of the large-subunit rRNAs (5S and 23S) as they appear in the ribosome. One of the protein subunits of the ribosome (more...)
In marked contrast to the central positions of the rRNA, the ribosomal proteins are generally located on the surface and fill in the gaps and crevices of the folded RNA (Figure 6-68). Some of these proteins contain globular domains on the ribosome surface that send out extended regions of polypeptide chain that penetrate short distances into holes in the RNA core (Figure 6-69). The main role of the ribosomal proteins seems to be to stabilize the RNA core, while permitting the changes in rRNA conformation that are necessary for this RNA to catalyze efficient protein synthesis.

Figure 6-68
Location of the protein components of the bacterial large ribosomal subunit. The rRNAs (5S and 23S) are depicted in gray and the large-subunit proteins (27 of the 31 total) in gold. For convenience, the protein structures depict only the polypeptide backbones. (more...)

Figure 6-69
Structure of the L15 protein in the large subunit of the bacterial ribosome. The globular domain of the protein lies on the surface of the ribosome and an extended region penetrates deeply into the RNA core of the ribosome. The L15 protein is shown in (more...)
Not only are the three binding sites for tRNAs (the A-, P-, and E-sites) on the ribosome formed primarily by the ribosomal RNAs, but the catalytic site for peptide bond formation is clearly formed by the 23S RNA, with the nearest amino acid located more than 1.8 nm away. This RNA-based catalytic site for peptidyl transferase is similar in many respects to those found in some proteins; it is a highly structured pocket that precisely orients the two reactants (the growing peptide chain and an aminoacyl-tRNA), and it provides a functional group to act as a general acid-base catalyst—in this case apparently, a ring nitrogen of adenine, instead of an amino acid side chain such as histidine (Figure 6-70). The ability of an RNA molecule to act as such a catalyst was initially surprising because RNA was thought to lack an appropriate chemical group that could both accept and donate a proton. Although the pK of adenine-ring nitrogens is usually around 3.5, the three-dimensional structure and charge distribution of the 23S rRNA active site force the pK of this apparently critical adenine into the neutral range and thereby create the enzymatic activity.

Figure 6-70
A possible reaction mechanism for the peptidyl transferase activity present in the large ribosomal subunit. The overall reaction is catalyzed by an active site in the 23S rRNA. In the first step of the proposed mechanism, the N3 of the active-site adenine (more...)
RNA molecules that possess catalytic activity are known as ribozymes. We saw earlier in this chapter how other ribozymes function in RNA-splicing reactions (for example, see Figure 6-36). In the final section of this chapter, we consider what the recently recognized ability of RNA molecules to function as catalysts for a wide variety of different reactions might mean for the early evolution of living cells. Here we need only note that there is good reason to suspect that RNA rather than protein molecules served as the first catalysts for living cells. If so, the ribosome, with its RNA core, might be viewed as a relic of an earlier time in life's history—when protein synthesis evolved in cells that were run almost entirely by ribozymes.
Nucleotide Sequences in mRNA Signal Where to Start Protein Synthesis
The initiation and termination of translation occur through variations on the translation elongation cycle described above. The site at which protein synthesis begins on the mRNA is especially crucial, since it sets the reading frame for the whole length of the message. An error of one nucleotide either way at this stage would cause every subsequent codon in the message to be misread, so that a nonfunctional protein with a garbled sequence of amino acids would result. The initiation step is also of great importance in another respect, since for most genes it is the last point at which the cell can decide whether the mRNA is to be translated and the protein synthesized; the rate of initiation thus determines the rate at which the protein is synthesized. We shall see in Chapter 7 that cells use several mechanisms to regulate translation initiation.
The translation of an mRNA begins with the codon AUG, and a special tRNA is required to initiate translation. This initiator tRNA always carries the amino acid methionine (in bacteria, a modified form of methionine—formylmethionine—is used) so that all newly made proteins have methionine as the first amino acid at their N-terminal end, the end of a protein that is synthesized first. This methionine is usually removed later by a specific protease. The initiator tRNA has a nucleotide sequence distinct from that of the tRNA that normally carries methionine.
In eucaryotes, the initiator tRNA (which is coupled to methionine) is first loaded into the small ribosomal subunit along with additional proteins called eucaryotic initiation factors, or eIFs (Figure 6-71). Of all the aminoacyl tRNAs in the cell, only the methionine-charged initiator tRNA is capable of tightly binding the small ribosome subunit without the complete ribosome present. Next, the small ribosomal subunit binds to the 5′ end of an mRNA molecule, which is recognized by virtue of its 5′ cap and its two bound initiation factors, eIF4E (which directly binds the cap) and eIF4G (see Figure 6-40). The small ribosomal subunit then moves forward (5′ to 3′) along the mRNA, searching for the first AUG. This movement is facilitated by additional initiation factors that act as ATP-powered helicases, allowing the small subunit to scan through RNA secondary structure. In 90% of mRNAs, translation begins at the first AUG encountered by the small subunit. At this point, the initiation factors dissociate from the small ribosomal subunit to make way for the large ribosomal subunit to assemble with it and complete the ribosome. The initiator tRNA is now bound to the P-site, leaving the A-site vacant. Protein synthesis is therefore ready to begin with the addition of the next aminoacyl tRNA molecule (see Figure 6-71).
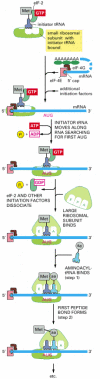
Figure 6-71
The initiation phase of protein synthesis in eucaryotes. Only three of the many translation initiation factors required for this process are shown. Efficient translation initiation also requires the poly-A tail of the mRNA bound by poly-A-binding proteins (more...)
The nucleotides immediately surrounding the start site in eucaryotic mRNAs influence the efficiency of AUG recognition during the above scanning process. If this recognition site is quite different from the consensus recognition sequence, scanning ribosomal subunits will sometimes ignore the first AUG codon in the mRNA and skip to the second or third AUG codon instead. Cells frequently use this phenomenon, known as "leaky scanning," to produce two or more proteins, differing in their N-termini, from the same mRNA molecule. It allows some genes to produce the same protein with and without a signal sequence attached at its N-terminus, for example, so that the protein is directed to two different compartments in the cell.
The mechanism for selecting a start codon in bacteria is different. Bacterial mRNAs have no 5′ caps to tell the ribosome where to begin searching for the start of translation. Instead, each bacterial mRNA contains a specific ribosome-binding site (called the Shine-Dalgarno sequence, named after its discoverers) that is located a few nucleotides upstream of the AUG at which translation is to begin. This nucleotide sequence, with the consensus 5′-AGGAGGU-3′, forms base pairs with the 16S rRNA of the small ribosomal subunit to position the initiating AUG codon in the ribosome. A set of translation initiation factors orchestrates this interaction, as well as the subsequent assembly of the large ribosomal subunit to complete the ribosome.
Unlike a eucaryotic ribosome, a bacterial ribosome can therefore readily assemble directly on a start codon that lies in the interior of an mRNA molecule, so long as a ribosome-binding site precedes it by several nucleotides. As a result, bacterial mRNAs are often polycistronic—that is, they encode several different proteins, each of which is translated from the same mRNA molecule (Figure 6-72). In contrast, a eucaryotic mRNA generally encodes only a single protein.

Figure 6-72
Structure of a typical bacterial mRNA molecule. Unlike eucaryotic ribosomes, which typically require a capped 5′ end, procaryotic ribosomes initiate transcription at ribosome-binding sites (Shine-Dalgarno sequences), which can be located anywhere (more...)
Stop Codons Mark the End of Translation
The end of the protein-coding message is signaled by the presence of one of three codons (UAA, UAG, or UGA) called stop codons (see Figure 6-50). These are not recognized by a tRNA and do not specify an amino acid, but instead signal to the ribosome to stop translation. Proteins known as release factors bind to any ribosome with a stop codon positioned in the A site, and this binding forces the peptidyl transferase in the ribosome to catalyze the addition of a water molecule instead of an amino acid to the peptidyl-tRNA (Figure 6-73). This reaction frees the carboxyl end of the growing polypeptide chain from its attachment to a tRNA molecule, and since only this attachment normally holds the growing polypeptide to the ribosome, the completed protein chain is immediately released into the cytoplasm. The ribosome then releases the mRNA and separates into the large and small subunits, which can assemble on another mRNA molecule to begin a new round of protein synthesis.

Figure 6-73
The final phase of protein synthesis. The binding of a release factor to an A-site bearing a stop codon terminates translation. The completed polypeptide is released and, after the action of a ribosome recycling factor (not shown), the ribosome dissociates (more...)
Release factors provide a dramatic example of molecular mimicry, whereby one type of macromolecule resembles the shape of a chemically unrelated molecule. In this case, the three-dimensional structure of release factors (made entirely of protein) bears an uncanny resemblance to the shape and charge distribution of a tRNA molecule (Figure 6-74). This shape and charge mimicry allows the release factor to enter the A-site on the ribosome and cause translation termination.
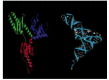
Figure 6-74
The structure of a human translation release factor (eRF1) and its resemblance to a tRNA molecule. The protein is on the left and the tRNA on the right. (From H. Song et al., Cell 100:311–321, 2000. © Elsevier.)
During translation, the nascent polypeptide moves through a large, water-filled tunnel (approximately 10 nm × 1.5 nm) in the large subunit of the ribosome (see Figure 6-68C). The walls of this tunnel, made primarily of 23S rRNA, are a patchwork of tiny hydrophobic surfaces embedded in a more extensive hydrophilic surface. This structure, because it is not complementary to any peptide structure, provides a "Teflon" coating through which a polypeptide chain can easily slide. The dimensions of the tunnel suggest that nascent proteins are largely unstructured as they pass through the ribosome, although some α-helical regions of the protein can form before leaving the ribosome tunnel. As it leaves the ribosome, a newly-synthesized protein must fold into its proper three-dimensional structure to be useful to the cell, and later in this chapter we discuss how this folding occurs. First, however, we review several additional aspects of the translation process itself.
Proteins Are Made on Polyribosomes
The synthesis of most protein molecules takes between 20 seconds and several minutes. But even during this very short period, multiple initiations usually take place on each mRNA molecule being translated. As soon as the preceding ribosome has translated enough of the nucleotide sequence to move out of the way, the 5′ end of the mRNA is threaded into a new ribosome. The mRNA molecules being translated are therefore usually found in the form of polyribosomes (also known as polysomes), large cytoplasmic assemblies made up of several ribosomes spaced as close as 80 nucleotides apart along a single mRNA molecule (Figure 6-75). These multiple initiations mean that many more protein molecules can be made in a given time than would be possible if each had to be completed before the next could start.

Figure 6-75
A polyribosome. (A) Schematic drawing showing how a series of ribosomes can simultaneously translate the same eucaryotic mRNA molecule. (B) Electron micrograph of a polyribosome from a eucaryotic cell. (B, courtesy of John Heuser.)
Both bacteria and eucaryotes utilize polysomes, and both employ additional strategies to speed up the rate of protein synthesis even further. Because bacterial mRNA does not need to be processed and is accessible to ribosomes while it is being made, ribosomes can attach to the free end of a bacterial mRNA molecule and start translating it even before the transcription of that RNA is complete, following closely behind the RNA polymerase as it moves along DNA. In eucaryotes, as we have seen, the 5′ and 3′ ends of the mRNA interact (see Figures 6-40 and 6-75A); therefore, as soon as a ribosome dissociates, its two subunits are in an optimal position to reinitiate translation on the same mRNA molecule.
Quality-Control Mechanisms Operate at Many Stages of Translation
Translation by the ribosome is a compromise between the opposing constraints of accuracy and speed. We have seen, for example, that the accuracy of translation (1 mistake per 104 amino acids joined) requires a time delay each time a new amino acid is added to a growing polypeptide chain, producing an overall speed of translation of 20 amino acids incorporated per second in bacteria. Mutant bacteria with a specific alteration in their small ribosomal subunit translate mRNA into protein with an accuracy considerably higher than this; however, protein synthesis is so slow in these mutants that the bacteria are barely able to survive.
We have also seen that attaining the observed accuracy of protein synthesis requires the expenditure of a great deal of free energy; this is expected, since, as discussed in Chapter 2, a price must be paid for any increase in order in the cell. In most cells, protein synthesis consumes more energy than any other biosynthetic process. At least four high-energy phosphate bonds are split to make each new peptide bond: two are consumed in charging a tRNA molecule with an amino acid (see Figure 6-56), and two more drive steps in the cycle of reactions occurring on the ribosome during synthesis itself (see Figure 6-66). In addition, extra energy is consumed each time that an incorrect amino acid linkage is hydrolyzed by a tRNA synthetase (see Figure 6-59) and each time that an incorrect tRNA enters the ribosome, triggers GTP hydrolysis, and is rejected (Figure 6-66). To be effective, these proofreading mechanisms must also remove an appreciable fraction of correct interactions; for this reason they are even more costly in energy than they might seem.
Other quality control mechanisms ensure that a eucaryotic mRNA molecule is complete before ribosomes even begin to translate it. Translating broken or partly processed mRNAs would be harmful to the cell, because truncated or otherwise aberrant proteins would be produced. In eucaryotes, we have seen that mRNA production involves not only transcription but also a series of elaborate RNA-processing steps; these take place in the nucleus, segregated from ribosomes, and only when the processing is complete are the mRNAs transported to the cytoplasm to be translated (see Figure 6-40). An mRNA molecule that was intact when it left the nucleus can, however, become broken in the cytosol. To avoid translating such broken mRNA molecules, the 5′ cap and the poly-A tail are both recognized by the translation-initiation apparatus before translation begins (see Figures 6-71 and 6-75).
Bacteria solve the problem of incomplete mRNAs in an entirely different way. Not only are there no signals at the 3′ ends of bacterial mRNAs, but also, as we have seen, translation often begins before the synthesis of the transcript has been completed. When the bacterial ribosome translates to the end of an incomplete RNA, a special RNA (called tmRNA) enters the A-site of the ribosome and is itself translated; this adds a special 11 amino acid tag to the C terminus of the truncated protein that signals to proteases that the entire protein is to be degraded (Figure 6-76).

Figure 6-76
The rescue of a bacterial ribosome stalled on an incomplete mRNA molecule. The tmRNA shown is a 363-nucleotide RNA with both tRNA and mRNA functions, hence its name. It carries an alanine and can enter the vacant A-site of a stalled ribosome to add this (more...)
There Are Minor Variations in the Standard Genetic Code
As discussed in Chapter 1, the genetic code (shown in Figure 6-50) applies to all three major branches of life, providing important evidence for the common ancestry of all life on Earth. Although rare, there are exceptions to this code, and we discuss some of them in this section. For example, Candida albicans, the most prevalent human fungal pathogen, translates the codon CUG as serine, whereas nearly all other organisms translate it as leucine. Mitochondria (which have their own genomes and encode much of their translational apparatus) also show several deviations from the standard code. For example, in mammalian mitochondria AUA is translated as methionine, whereas in the cytosol of the cell it is translated as isoleucine (see Table 14-3, p. 814).
The type of deviation in the genetic code discussed above is "hardwired" into the organisms or the organelles in which it occurs. A different type of variation, sometimes called translational recoding, occurs in many cells. In this case, other nucleotide sequence information present in an mRNA can change the meaning of the genetic code at a particular site in the mRNA molecule. The standard code allows cells to manufacture proteins using only 20 amino acids. However, bacteria, archaea, and eucaryotes have available to them a twenty-first amino acid that can be incorporated directly into a growing polypeptide chain through translational recoding. Selenocysteine, which is essential for the efficient function of a variety of enzymes, contains a selenium atom in place of the sulfur atom of cysteine. Selenocysteine is produced from a serine attached to a special tRNA molecule that base-pairs with the UGA codon, a codon normally used to signal a translation stop. The mRNAs for proteins in which selenocysteine is to be inserted at a UGA codon carry an additional nucleotide sequence in the mRNA nearby that causes this recoding event (Figure 6-77).

Figure 6-77
Incorporation of selenocysteine into a growing polypeptide chain. A specialized tRNA is charged with serine by the normal seryl-tRNA synthetase, and the serine is subsequently converted enzymatically to selenocysteine. A specific RNA structure in the (more...)
Another form of recoding is translational frameshifting. This type of recoding is commonly used by retroviruses, a large group of eucaryotic viruses, in which it allows more than one protein to be synthesized from a single mRNA. These viruses commonly make both the capsid proteins (Gag proteins) and the viral reverse transcriptase and integrase (Pol proteins) from the same RNA transcript (see Figure 5-73). Such a virus needs many more copies of the Gag proteins than it does of the Pol proteins, and they achieve this quantitative adjustment by encoding the pol genes just after the gag genes but in a different reading frame. A stop codon at the end of the gag coding sequence can be bypassed on occasion by an intentional translational frameshift that occurs upstream of it. This frameshift occurs at a particular codon in the mRNA and requires a specific recoding signal, which seems to be a structural feature of the RNA sequence downstream of this site (Figure 6-78).

Figure 6-78
The translational frameshifting that produces the reverse transcriptase and integrase of a retrovirus. The viral reverse transcriptase and integrase are produced by proteolytic processing of a large protein (the Gag-Pol fusion protein) consisting of both (more...)
Many Inhibitors of Procaryotic Protein Synthesis Are Useful as Antibiotics
Many of the most effective antibiotics used in modern medicine are compounds made by fungi that act by inhibiting bacterial protein synthesis. Some of these drugs exploit the structural and functional differences between bacterial and eucaryotic ribosomes so as to interfere preferentially with the function of bacterial ribosomes. Thus some of these compounds can be taken in high doses without undue toxicity to humans. Because different antibiotics bind to different regions of bacterial ribosomes, they often inhibit different steps in the synthetic process. Some of the more common antibiotics of this kind are listed in Table 6-3 along with several other inhibitors of protein synthesis, some of which act on eucaryotic cells and therefore cannot be used as antibiotics.
Table 6-3
Inhibitors of Protein or RNA Synthesis.
Because they block specific steps in the processes that lead from DNA to protein, many of the compounds listed in Table 6-3 are useful for cell biological studies. Among the most commonly used drugs in such experimental studies are chloramphenicol, cycloheximide, and puromycin, all of which specifically inhibit protein synthesis. In a eucaryotic cell, for example, chloramphenicol inhibits protein synthesis on ribosomes only in mitochondria (and in chloroplasts in plants), presumably reflecting the procaryotic origins of these organelles (discussed in Chapter 14). Cycloheximide, in contrast, affects only ribosomes in the cytosol. Puromycin is especially interesting because it is a structural analog of a tRNA molecule linked to an amino acid and is therefore another example of molecular mimicry; the ribosome mistakes it for an authentic amino acid and covalently incorporates it at the C-terminus of the growing peptide chain, thereby causing the premature termination and release of the polypeptide. As might be expected, puromycin inhibits protein synthesis in both procaryotes and eucaryotes.
Having described the translation process itself, we now discuss how its products—the proteins of the cell—fold into their correct three-dimensional conformations.
A Protein Begins to Fold While It Is Still Being Synthesized
The process of gene expression is not over when the genetic code has been used to create the sequence of amino acids that constitutes a protein. To be useful to the cell, this new polypeptide chain must fold up into its unique three-dimensional conformation, bind any small-molecule cofactors required for its activity, be appropriately modified by protein kinases or other protein-modifying enzymes, and assemble correctly with the other protein subunits with which it functions (Figure 6-79).
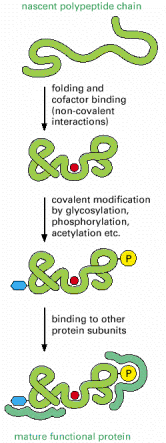
Figure 6-79
Steps in the creation of a functional protein. As indicated, translation of an mRNA sequence into an amino acid sequence on the ribosome is not the end of the process of forming a protein. To be useful to the cell, the completed polypeptide chain must (more...)
The information needed for all of the protein maturation steps listed above is ultimately contained in the sequence of linked amino acids that the ribosome produces when it translates an mRNA molecule into a polypeptide chain. As discussed in Chapter 3, when a protein folds into a compact structure, it buries most of its hydrophobic residues in an interior core. In addition, large numbers of noncovalent interactions form between various parts of the molecule. It is the sum of all of these energetically favorable arrangements that determines the final folding pattern of the polypeptide chain—as the conformation of lowest free energy (see p. 134).
Through many millions of years of evolutionary time, the amino acid sequence of each protein has been selected not only for the conformation that it adopts but also for an ability to fold rapidly, as its polypeptide chain spins out of the ribosome starting from the N-terminal end. Experiments have demonstrated that once a protein domain in a multi-domain protein emerges from the ribosome, it forms a compact structure within a few seconds that contains most of the final secondary structure (α helices and β sheets) aligned in roughly the right way (Figure 6-80). For many protein domains, this unusually open and flexible structure, which is called a molten globule, is the starting point for a relatively slow process in which many side-chain adjustments occur that eventually form the correct tertiary structure. Nevertheless, because it takes several minutes to synthesize a protein of average size, a great deal of the folding process is complete by the time the ribosome releases the C-terminal end of a protein (Figure 6-81).

Figure 6-80
The structure of a molten globule. (A) A molten globule form of cytochrome b 562 is more open and less highly ordered than the final folded form of the protein, shown in (B). Note that the molten globule contains most of the secondary structure of the (more...)
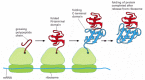
Figure 6-81
The co-translational folding of a protein. A growing polypeptide chain is shown acquiring its secondary and tertiary structure as it emerges from a ribosome. The N-terminal domain folds first, while the C-terminal domain is still being synthesized. In (more...)
Molecular Chaperones Help Guide the Folding of Many Proteins
The folding of many proteins is made more efficient by a special class of proteins called molecular chaperones. The latter proteins are useful for cells because there are a variety of different paths that can be taken to convert the molten globule form of a protein to the protein's final compact conformation. For many proteins, some of the intermediates formed along the way would aggregate and be left as off-pathway dead ends without the intervention of a chaperone that resets the folding process (Figure 6-82).

Figure 6-82
A current view of protein folding. Each domain of a newly synthesized protein rapidly attains a "molten globule" state. Subsequent folding occurs more slowly and by multiple pathways, often involving the help of a molecular chaperone. (more...)
Molecular chaperones were first identified in bacteria when E. coli mutants that failed to allow bacteriophage lambda to replicate in them were studied. These mutant cells produce slightly altered versions of the chaperone machinery, and as a result they are defective in specific steps in the assembly of the viral proteins. The molecular chaperones are included among the heat-shock proteins (hence their designation as hsp), because they are synthesized in dramatically increased amounts after a brief exposure of cells to an elevated temperature (for example, 42°C for cells that normally live at 37°C). This reflects the operation of a feedback system that responds to any increase in misfolded proteins (such as those produced by elevated temperatures) by boosting the synthesis of the chaperones that help these proteins refold.
Eucaryotic cells have at least two major families of molecular chaperones—known as the hsp60 and hsp70 proteins. Different family members function in different organelles. Thus, as discussed in Chapter 12, mitochondria contain their own hsp60 and hsp70 molecules that are distinct from those that function in the cytosol, and a special hsp70 (called BIP) helps to fold proteins in the endoplasmic reticulum.
The hsp60-like and hsp70 proteins each work with their own small set of associated proteins when they help other proteins to fold. They share an affinity for the exposed hydrophobic patches on incompletely folded proteins, and they hydrolyze ATP, often binding and releasing their protein with each cycle of ATP hydrolysis. In other respects, the two types of hsp proteins function differently. The hsp70 machinery acts early in the life of many proteins, binding to a string of about seven hydrophobic amino acids before the protein leaves the ribosome (Figure 6-83). In contrast, hsp60-like proteins form a large barrel-shaped structure that acts later in a protein's life, after it has been fully synthesized. This type of chaperone forms an "isolation chamber" into which misfolded proteins are fed, preventing their aggregation and providing them with a favorable environment in which to attempt to refold (Figure 6-84).
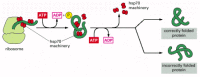
Figure 6-83
The hsp70 family of molecular chaperones. These proteins act early, recognizing a small stretch of hydrophobic amino acids on a protein's surface. Aided by a set of smaller hsp40 proteins, an hsp70 monomer binds to its target protein and then hydrolyzes (more...)
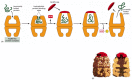
Figure 6-84
The structure and function of the hsp60 family of molecular chaperones. (A) The catalysis of protein refolding. As indicated, a misfolded protein is initially captured by hydrophobic interactions along one rim of the barrel. The subsequent binding of (more...)
Exposed Hydrophobic Regions Provide Critical Signals for Protein Quality Control
If radioactive amino acids are added to cells for a brief period, the newly synthesized proteins can be followed as they mature into their final functional form. It is this type of experiment that shows that the hsp70 proteins act first, beginning when a protein is still being synthesized on a ribosome, and that the hsp60-like proteins are called into play only later to help in folding completed proteins. However, the same experiments reveal that only a subset of the newly synthesized proteins becomes involved: perhaps 20% of all proteins with the hsp70 and 10% with the hsp60-like molecular chaperones. How are these proteins selected for this ATP-catalyzed refolding?
Before answering, we need to pause to consider the post-translational fate of proteins more broadly. A protein that has a sizable exposed patch of hydrophobic amino acids on its surface is usually abnormal: it has either failed to fold correctly after leaving the ribosome, suffered an accident that partly unfolded it at a later time, or failed to find its normal partner subunit in a larger protein complex. Such a protein is not merely useless to the cell, it can be dangerous. Many proteins with an abnormally exposed hydrophobic region can form large aggregates, precipitating out of solution. We shall see that, in rare cases, such aggregates do form and cause severe human diseases. But in the vast majority of cells, powerful protein quality control mechanisms prevent such disasters.
Given this background, it is not surprising that cells have evolved elaborate mechanisms that recognize and remove the hydrophobic patches on proteins. Two of these mechanisms depend on the molecular chaperones just discussed, which bind to the patch and attempt to repair the defective protein by giving it another chance to fold. At the same time, by covering the hydrophobic patches, these chaperones transiently prevent protein aggregation. Proteins that very rapidly fold correctly on their own do not display such patches and are therefore bypassed by chaperones.
Figure 6-85 outlines all of the quality control choices that a cell makes for a difficult-to-fold, newly synthesized protein. As indicated, when attempts to refold a protein fail, a third mechanism is called into play that completely destroys the protein by proteolysis. The proteolytic pathway begins with the recognition of an abnormal hydrophobic patch on a protein's surface, and it ends with the delivery of the entire protein to a protein destruction machine, a complex protease known as the proteasome. As described next, this process depends on an elaborate protein-marking system that also carries out other central functions in the cell by destroying selected normal proteins.

Figure 6-85
The cellular mechanisms that monitor protein quality after protein synthesis. As indicated, a newly synthesized protein sometimes folds correctly and assembles with its partners on its own, in which case it is left alone. Incompletely folded proteins (more...)
The Proteasome Degrades a Substantial Fraction of the Newly Synthesized Proteins in Cells
Cells quickly remove the failures of their translation processes. Recent experiments suggest that as many as one-third of the newly made polypeptide chains are selected for rapid degradation as a result of the protein quality control mechanisms just described. The final disposal apparatus in eucaryotes is the proteasome, an abundant ATP-dependent protease that constitutes nearly 1% of cellular protein. Present in many copies dispersed throughout the cytosol and the nucleus, the proteasome also targets proteins of the endoplasmic reticulum (ER): those proteins that fail either to fold or to be assembled properly after they enter the ER are detected by an ER-based surveillance system that retrotranslocate them back to the cytosol for degradation (discussed in Chapter 12).
Each proteasome consists of a central hollow cylinder (the 20S core proteasome) formed from multiple protein subunits that assemble as a cylindrical stack of four heptameric rings. Some of these subunits are distinct proteases whose active sites face the cylinder's inner chamber (Figure 6-86A). Each end of the cylinder is normally associated with a large protein complex (the 19S cap) containing approximately 20 distinct polypeptides (Figure 6-86B). The cap subunits include at least six proteins that hydrolyze ATP; located near the edge of the cylinder, these ATPases are thought to unfold the proteins to be digested and move them into the interior chamber for proteolysis. A crucial property of the proteasome, and one reason for the complexity of its design, is the processivity of its mechanism: in contrast to a "simple" protease that cleaves a substrate's polypeptide chain just once before dissociating, the proteasome keeps the entire substrate bound until all of it is converted into short peptides.
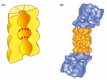
Figure 6-86
The proteasome. (A) A cut-away view of the structure of the central 20S cylinder, as determined by x-ray crystallography, with the active sites of the proteases indicated by red dots. (B) The structure of the entire proteasome, in which the central cylinder (more...)
The 19S caps act as regulated "gates" at the entrances to the inner proteolytic chamber, being also responsible for binding a targeted protein substrate to the proteasome. With a few exceptions, the proteasomes act on proteins that have been specifically marked for destruction by the covalent attachment of multiple copies of a small protein called ubiquitin (Figure 6-87A). Ubiquitin exists in cells either free or covalently linked to a huge variety of intracellular proteins. For most of these proteins, this tagging by ubiquitin results in their destruction by the proteasome.

Figure 6-87
Ubiquitin and the marking of proteins with multiubiquitin chains. (A) The three-dimensional structure of ubiquitin; this relatively small protein contains 76 amino acids. (B) The C-terminus of ubiquitin is initially activated through its high-energy thioester (more...)
An Elaborate Ubiquitin-conjugating System Marks Proteins for Destruction
Ubiquitin is prepared for conjugation to other proteins by the ATP-dependent ubiquitin-activating enzyme (E1), which creates an activated ubiquitin that is transferred to one of a set of ubiquitin-conjugating (E2) enzymes. The E2 enzymes act in conjunction with accessory (E3) proteins. In the E2-E3 complex, called ubiquitin ligase, the E3 component binds to specific degradation signals in protein substrates, helping E2 to form a multiubiquitin chain linked to a lysine of the substrate protein. In this chain, the C-terminal residue of each ubiquitin is linked to a specific lysine of the preceding ubiquitin molecule, producing a linear series of ubiquitin-ubiquitin conjugates (Figure 6-87B). It is this multiubiquitin chain on a target protein that is recognized by a specific receptor in the proteasome.
There are roughly 30 structurally similar but distinct E2 enzymes in mammals, and hundreds of different E3 proteins that form complexes with specific E2 enzymes. The ubiquitin-proteasome system thus consists of many distinct but similarly organized proteolytic pathways, which have in common both the E1 enzyme at the "top" and the proteasome at the "bottom," and differ by the compositions of their E2-E3 ubiquitin ligases and accessory factors. Distinct ubiquitin ligases recognize different degradation signals, and therefore target for degradation distinct subsets of intracellular proteins that bear these signals.
Denatured or otherwise misfolded proteins, as well as proteins containing oxidized or other abnormal amino acids, are recognized and destroyed because abnormal proteins tend to present on their surface amino acid sequences or conformational motifs that are recognized as degradation signals by a set of E3 molecules in the ubiquitin-proteasome system; these sequences must of course be buried and therefore inaccessible in the normal counterparts of these proteins. However, a proteolytic pathway that recognizes and destroys abnormal proteins must be able to distinguish between completed proteins that have "wrong" conformations and the many growing polypeptides on ribosomes (as well as polypeptides just released from ribosomes) that have not yet achieved their normal folded conformation. This is not a trivial problem; the ubiquitin-proteasome system is thought to destroy some of the nascent and newly formed protein molecules not because these proteins are abnormal as such but because they transiently expose degradation signals that are buried in their mature (folded) state.
Many Proteins Are Controlled by Regulated Destruction
One function of intracellular proteolytic mechanisms is to recognize and eliminate misfolded or otherwise abnormal proteins, as just described. Yet another function of these proteolytic pathways is to confer short half-lives on specific normal proteins whose concentrations must change promptly with alterations in the state of a cell. Some of these short-lived proteins are degraded rapidly at all times, while many others are conditionally short-lived, that is, they are metabolically stable under some conditions, but become unstable upon a change in the cell's state. For example, mitotic cyclins are long-lived throughout the cell cycle until their sudden degradation at the end of mitosis, as explained in Chapter 17.
How is such a regulated destruction of a protein controlled? A variety of mechanisms are known, as illustrated through specific examples later in this book. In one general class of mechanism (Figure 6-88A), the activity of a ubiquitin ligase is turned on either by E3 phosphorylation or by an allosteric transition in an E3 protein caused by its binding to a specific small or large molecule. For example, the anaphase-promoting complex (APC) is a multisubunit ubiquitin ligase that is activated by a cell-cycle-timed subunit addition at mitosis. The activated APC then causes the degradation of mitotic cyclins and several other regulators of the metaphase-anaphase transition (see Figure 17-20).
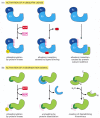
Figure 6-88
Two general ways of inducing the degradation of a specific protein. (A) Activation of a specific E3 molecule creates a new ubiquitin ligase. (B) Creation of an exposed degradation signal in the protein to be degraded. This signal binds a ubiquitin ligase, (more...)
Alternatively, in response either to intracellular signals or to signals from the environment, a degradation signal can be created in a protein, causing its rapid ubiquitylation and destruction by the proteasome. One common way to create such a signal is to phosphorylate a specific site on a protein that unmasks a normally hidden degradation signal. Another way to unmask such a signal is by the regulated dissociation of a protein subunit. Finally, powerful degradation signals can be created by a single cleavage of a peptide bond, provided that this cleavage creates a new N-terminus that is recognized by a specific E3 as a "destabilizing" N-terminal residue (Figure 6-88B).
The N-terminal type of degradation signal arises because of the "N-end rule," which relates the half-life of a protein in vivo to the identity of its N-terminal residue. There are 12 destabilizing residues in the N-end rule of the yeast S. cerevisiae (Arg, Lys, His, Phe, Leu, Tyr, Trp, Ile, Asp, Glu, Asn, and Gln), out of the 20 standard amino acids. The destabilizing N-terminal residues are recognized by a special ubiquitin ligase that is conserved from yeast to humans.
As we have seen, all proteins are initially synthesized bearing methionine (or formylmethionine in bacteria), as their N-terminal residue, which is a stabilizing residue in the N-end rule. Special proteases, called methionine aminopeptidases, will often remove the first methionine of a nascent protein, but they will do so only if the second residue is also stabilizing in the yeast-type N-end rule. Therefore, it was initially unclear how N-end rule substrates form in vivo. However, it has recently been discovered that a subunit of cohesin, a protein complex that holds sister chromatids together, is cleaved by a site-specific protease at the metaphase-anaphase transition. This cell-cycle-regulated cleavage allows separation of the sister chromatids and leads to the completion of mitosis (see Figure 17-26). The C-terminal fragment of the cleaved subunit bears an N-terminal arginine, a destabilizing residue in the N-end rule. Mutant cells lacking the N-end rule pathway exhibit a greatly increased frequency of chromosome loss, presumably because a failure to degrade this fragment of the cohesin subunit interferes with the formation of new chromatid-associated cohesin complexes in the next cell cycle.
Abnormally Folded Proteins Can Aggregate to Cause Destructive Human Diseases
When all of a cell's protein quality controls fail, large protein aggregates tend to accumulate in the affected cell (see Figure 6-85). Some of these aggregates, by adsorbing critical macromolecules to them, can severely damage cells and even cause cell death. The protein aggregates released from dead cells tend to accumulate in the extracellular matrix that surrounds the cells in a tissue, and in extreme cases they can also damage tissues. Because the brain is composed of a highly organized collection of nerve cells, it is especially vulnerable. Not surprisingly, therefore, protein aggregates primarily cause diseases of neuro-degeneration. Prominent among these are Huntington's disease and Alzheimer's disease—the latter causing age-related dementia in more than 20 million people in today's world.
For a particular type of protein aggregate to survive, grow, and damage an organism, it must be highly resistant to proteolysis both inside and outside the cell. Many of the protein aggregates that cause problems form fibrils built from a series of polypeptide chains that are layered one over the other as a continuous stack of β sheets. This so-called cross-beta filament (Figure 6-89C) tends to be highly resistant to proteolysis. This resistance presumably explains why this structure is observed in so many of the neurological disorders caused by protein aggregates, where it produces abnormally staining deposits known as amyloid.

Figure 6-89
Protein aggregates that cause human disease. (A) Schematic illustration of the type of conformational change in a protein that produces material for a cross-beta filament. (B) Diagram illustrating the self-infectious nature of the protein aggregation (more...)
One particular variety of these diseases has attained special notoriety. These are the prion diseases. Unlike Huntington's or Alzheimer's disease, a prion disease can spread from one organism to another, providing that the second organism eats a tissue containing the protein aggregate. A set of diseases—called scrapie in sheep, Creutzfeldt-Jacob disease (CJD) in humans, and bovine spongiform encephalopathy (BSE) in cattle—are caused by a misfolded, aggregated form of a protein called PrP (for prion protein). The PrP is normally located on the outer surface of the plasma membrane, most prominently in neurons. Its normal function is not known. However, PrP has the unfortunate property of being convertible to a very special abnormal conformation (Figure 6-89A). This conformation not only forms protease-resistant, cross-beta filaments; it also is "infectious" because it converts normally folded molecules of PrP to the same form. This property creates a positive feedback loop that propagates the abnormal form of PrP, called PrP* (Figure 6-89B) and thereby allows PrP to spread rapidly from cell to cell in the brain, causing the death of both animals and humans. It can be dangerous to eat the tissues of animals that contain PrP*, as witnessed most recently by the spread of BSE (commonly referred to as the "mad cow disease") from cattle to humans in Great Britain.
Fortunately, in the absence of PrP*, PrP is extraordinarily difficult to convert to its abnormal form. Although very few proteins have the potential to misfold into an infectious conformation, a similar transformation has been discovered to be the cause of an otherwise mysterious "protein-only inheritance" observed in yeast cells.
There Are Many Steps From DNA to Protein
We have seen so far in this chapter that many different types of chemical reactions are required to produce a properly folded protein from the information contained in a gene (Figure 6-90). The final level of a properly folded protein in a cell therefore depends upon the efficiency with which each of the many steps is performed.

Figure 6-90
The production of a protein by a eucaryotic cell. The final level of each protein in a eucaryotic cell depends upon the efficiency of each step depicted.
We discuss in Chapter 7 that cells have the ability to change the levels of their proteins according to their needs. In principle, any or all of the steps in Figure 6-90) could be regulated by the cell for each individual protein. However, as we shall see in Chapter 7, the initiation of transcription is the most common point for a cell to regulate the expression of each of its genes. This makes sense, inasmuch as the most efficient way to keep a gene from being expressed is to block the very first step—the transcription of its DNA sequence into an RNA molecule.
Summary
The translation of the nucleotide sequence of an mRNA molecule into protein takes place in the cytoplasm on a large ribonucleoprotein assembly called a ribosome. The amino acids used for protein synthesis are first attached to a family of tRNA molecules, each of which recognizes, by complementary base-pair interactions, particular sets of three nucleotides in the mRNA (codons). The sequence of nucleotides in the mRNA is then read from one end to the other in sets of three according to the genetic code.
To initiate translation, a small ribosomal subunit binds to the mRNA molecule at a start codon (AUG) that is recognized by a unique initiator tRNA molecule. A large ribosomal subunit binds to complete the ribosome and begin the elongation phase of protein synthesis. During this phase, aminoacyl tRNAs—each bearing a specific amino acid bind sequentially to the appropriate codon in mRNA by forming complementary base pairs with the tRNA anticodon. Each amino acid is added to the C-terminal end of the growing polypeptide by means of a cycle of three sequential steps: aminoacyl-tRNA binding, followed by peptide bond formation, followed by ribosome translocation. The mRNA molecule progresses codon by codon through the ribosome in the 5′-to-3′ direction until one of three stop codons is reached. A release factor then binds to the ribosome, terminating translation and releasing the completed polypeptide.
Eucaryotic and bacterial ribosomes are closely related, despite differences in the number and size of their rRNA and protein components. The rRNA has the dominant role in translation, determining the overall structure of the ribosome, forming the binding sites for the tRNAs, matching the tRNAs to codons in the mRNA, and providing the peptidyl transferase enzyme activity that links amino acids together during translation.
In the final steps of protein synthesis, two distinct types of molecular chaperones guide the folding of polypeptide chains. These chaperones, known as hsp60 and hsp70, recognize exposed hydrophobic patches on proteins and serve to prevent the protein aggregation that would otherwise compete with the folding of newly synthesized proteins into their correct three-dimensional conformations. This protein folding process must also compete with a highly elaborate quality control mechanism that destroys proteins with abnormally exposed hydrophobic patches. In this case, ubiquitin is covalently added to a misfolded protein by a ubiquitin ligase, and the resulting multiubiquitin chain is recognized by the cap on a proteasome to move the entire protein to the interior of the proteasome for proteolytic degradation. A closely related proteolytic mechanism, based on special degradation signals recognized by ubiquitin ligases, is used to determine the lifetime of many normally folded proteins. By this method, selected normal proteins are removed from the cell in response to specific signals.
Where Do the Instructions for Building a Protein Come From Originally?
Source: https://www.ncbi.nlm.nih.gov/books/NBK26829/
0 Response to "Where Do the Instructions for Building a Protein Come From Originally?"
Post a Comment